 Cardiopatías Congénitas
Cardiopatías CongénitasEsta cardiopatía consiste en la hipoplasia de todo el corazón izquierdo, y algunos la denominan Síndrome del Corazón Izquierdo Hipoplásico (SCIH). Es posiblemente la cardiopatía más grave y de peor pronóstico, aunque en los últimos años hay diferentes opciones terapéuticas.
El SCIH representa el 1.4-3.8% de todas las cardiopatías congénitas. A pesar de su baja incidencia, 0.016-0.036 % de recién nacidos vivos, el SCIH es la causa del 23% de mortalidad cardiológica en la primera semana de vida, y el 15% en el primer mes de vida. Existe una predominancia masculina para el SCIH (55-67%). El riesgo de recurrencia en familias con un hijo afectado es 0.5-2%. Adicionalmente, el riesgo de otras formas de cardiopatía congénita en familias con un hijo afectado es 2.2-13.5%. Aunque probablemente la modalidad de herencia sea multifactorial, el riesgo de recurrencia entre hermanos sugiere una vía autosómica recesiva de transmisión. Entre un 15-30% de los pacientes con diagnóstico de SCIH asocia anomalías extracardiacas (hernia diafragmática, atresia duodenal, atresia de vías biliares, malrotación, onfalocele y fibrosis quística) y síndromes genéticos (Turner, Noonan, Smith-Lemli-Opitz, Holt-Oram, Ellis-van Creveld y CHARGE). Se han identificado varias anomalías cromosómicas relacionadas con el SCIH, como Trisomía 13, Trisomía 18, Trisomía 21, duplicación del brazo corto del cromosoma 12, translocación 4q-, 4p-, 7q-, 11q, duplicaciones de 16q- y 18p-. La investigación en curso de genética molecular podría permitir en un futuro cercano un mejor conocimiento de las causas del SCIH, como son las mutaciones en reguladores de transcripción y señalización, entre las que se encuentran la secuencia NOTCH1 y las proteínas HRT.
Circulación fetal en el SCIH

En la imagen se observa una imagen de cuatro cámaras del SCIH durante el periodo fetal. A la izquierda puede verse el ventrículo izquierdo (VI) hipoplásico que no se ve y a la derecha el flujo en color a través de la válvula tricúspide entre aurícula derecha (AD) y ventrículos derecho (VD), y un mínimo flujo anterógrado que entra en el VI desde aurícula izquierda (AI).
Se ha objetivado que los fetos con SCIH tienen un foramen oval más pequeño que los fetos con corazones normales. Hay algunos, incluso, sin foramen oval. A través del foramen oval, el VI recibe sangre, siendo ésta esencial para su desarrollo. Otras anomalías anatómicas asociadas son alteraciones genéticas de las válvulas (mitral o aórtica, como la aorta bicúspide). La circulación sanguínea de estos fetos no es normal: La sangre de la AI pasa por la CIA a la AD y VD. Desde aquí pasa la sangre anterógradamente a la arteria pulmonar, al ductus, aorta descendente hacia todo el cuerpo y las piernas, y de forma retrógrada hacia la cabeza. Muy poca sangre pasa a los pulmones y, por tanto, muy poca regresa a la aurícula izquierda y muy poca o nula al VI, motivo por el que éste no se desarrolla. Se ha contemplado también el intervencionismo fetal en esta patología, con la esperanza de propiciar un mejor desarrollo del VI.
Anatomía y fisiopatología
Como en realidad no hay un VI normal, el corazón de estos niños funciona como si sólo tuviera medio corazón, es decir, sólo un ventrículo, el ventrículo derecho (VD) (corazón univentricular). Para que vivan estos niños, el VD pasa a ser la bomba principal del corazón, y además de enviar la sangre no oxigenada a los pulmones (que es la función normal del VD), asume también la función del VI, que es la de enviar la sangre a todo el cuerpo. Esto significa que la sangre oxigenada (roja) que llega de las venas pulmonares (VP) a la aurícula izquierda (AI), no puede pasar hacia el VI y la aorta como sería lo normal, porque la válvula mitral puede ser hipoplásica o no existir (atresia mitral), el VI muy pequeño o hipoplásico y la válvula aórtica muy pequeña y estenótica o no existir (atresia aórtica); debe necesariamente pasar a la aurícula derecha (AD) a través de una comunicación interauricular (CIA), mezclándose completamente con la sangre no oxigenada (azul) procedente de las cavas (VC). Ambas sangres mezcladas (violeta) pasan al VD y de éste a la arteria pulmonar (AP). En la AP, la sangre se bifurca en dos circulaciones: 1) Hacia los pulmones (P), para su oxigenación, que es la función normal del VD, y 2) Hacia la aorta y todo el cuerpo (C) a través del ductus abierto, que es la función del VI. Es esencial que el ductus se mantenga abierto después del nacimiento del niño; si se cierra, el niño fallece. En ambas circulaciones, pulmonar y aórtica, la sangre está mezclada (violeta), de forma que el sistema es poco eficiente, ya que parte de la sangre que llega al pulmón llega inútilmente, pues ya está oxigenada, y parte de la sangre que llega a todo el cuerpo llega también inútilmente, pues no está correctamente oxigenada. Para mantener el nivel de oxigenación adecuado para todo el cuerpo, el corazón tiene que bombear más sangre de lo normal y trabajar más. Pero sabemos que el VD, que asume las funciones de ambos ventrículos (VD y VI), tiene una estructura sólo adecuada para trabajo de poca entidad, como es bombear sangre a los pulmones, pero carece de la estructura muscular del VI, que es la adecuada para llevar a cabo trabajos intensos, como bombear sangre a todo el cuerpo; es fácil que el VD sucumba al asumir la función de ambos ventrículos.
Es esencial que el ductus y la CIA se mantengan abiertos para que sobreviva el niño. Incluso con ambas estructuras abiertas no es fácil la evolución de estos niños, pues estando conectado el sistema pulmonar al aórtico (a través del ductus), la sangre cursa hacia el sistema de menor resistencia al flujo, el pulmonar, pudiendo aparecer hiperaflujo pulmonar, por un lado y, por otro lado, bajo gasto sistémico, una situación que resulta engañosa clínicamente, dando la impresión de que el niño está bien oxigenado, cuando en realidad está próximo al shock cardiaco y al fallo multiorgánico por bajo gasto. Si la CIA es restrictiva o pequeña, la sangre se estanca en la aurícula izquierda y pulmones, frenando la llegada de sangre oxigenada a los pulmones y causando hipoxemia.
El espectro de malformaciones del SCIH incluye: Atresia valvular aórtica con atresia mitral (36-46%); Atresia valvular aórtica con diferentes grados de estenosis mitral (20-29%); y estenosis aórtica con estenosis mitral (23-26%), además de un VI poco desarrollado y la existencia obligada de un ductus permeable y una CIA.
Enfoque diagnóstico

La ecocardiografía 2D será suficiente para el diagnóstico del SCIH. En la en la imagen de la izquierda podemos observar que el VI es hipoplásico, globuloso e hiper-ecogénico, mucho más pequeño que el VD. En la imagen de la derecha se muestra un arco aórtico con flujo retrógrado (color rojo). La imagen ecográfica determinará todos los detalles anatómicos incluido la presencia o no de insuficiencia tricúspide y la dirección de flujo en el ductus durante la diástole.
El cateterismo cardiaco de rutina no será necesario para la evaluación neonatal del SCIH.
Tratamiento

En el dibujo adjunto observamos la pequeñez del VI y de la aorta y un ductus grande permeable y una CIA. El tratamiento es complejo y se lleva a cabo en varios estadios:
Estadío-1 de Paliación
Manejo Preoperatorio: Los neonatos con SCIH requerirán infusión continua de prostaglandinas E1 (PGE) para mantener el ductus permeable. Si no existe CIA o ésta es muy restrictiva, son necesarios su apertura y ensanche por métodos quirúrgicos o percutáneos. Aquellos niños que presentan shock cardiogénico necesitan inmediata resucitación, y a menudo requieren intubación, expansión volumétrica y soporte inotrópico. Las situaciones de hiperaflujo pulmonar, si no se controlan con manejo conservador médico, deben conducir a una pronta cirugía.
El momento óptimo para realizar la corrección quirúrgica no es conocido, y será específico de cada centro. Sin embargo, después del primer mes de vida, el riesgo aumenta. En este estadío hay varias estrategias quirúrgicas:
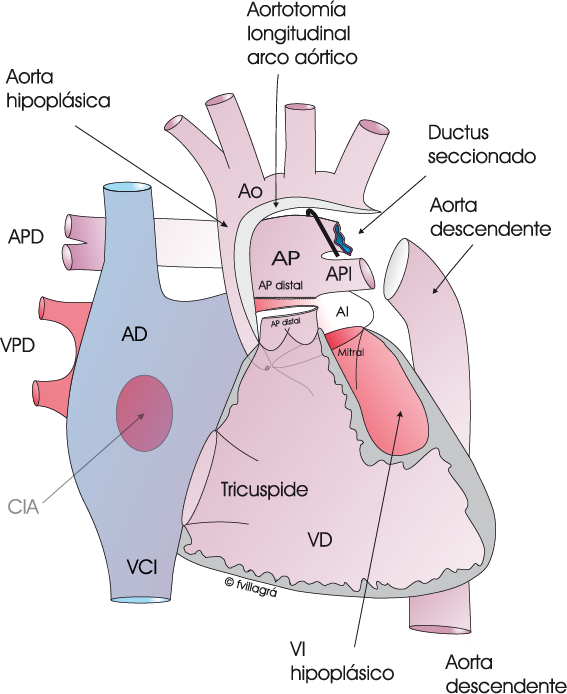
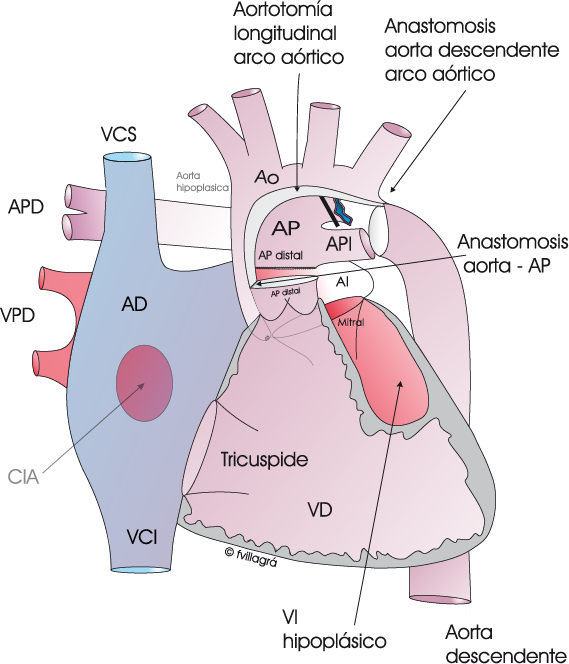
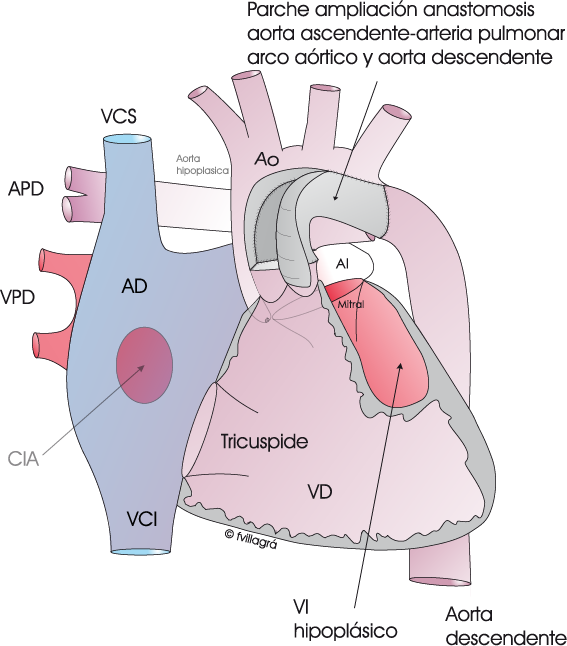
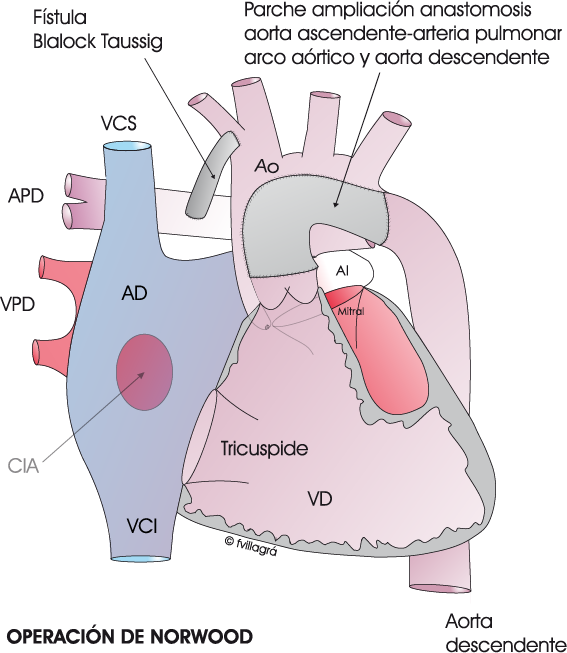
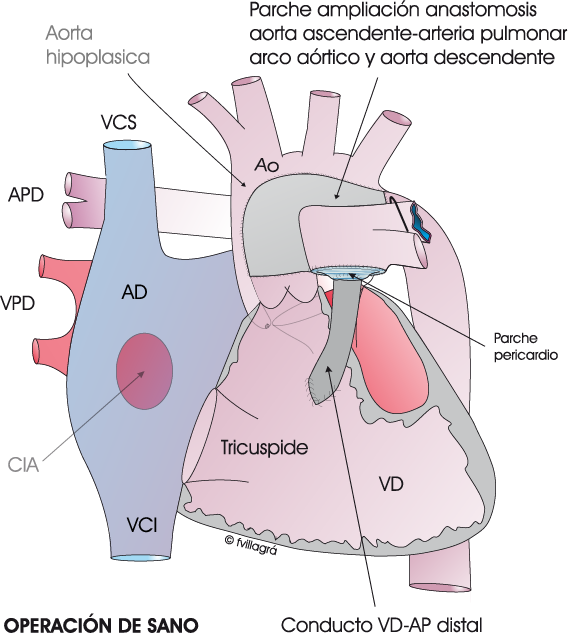
El procedimiento de Norwood “Clásico” o Norwood Sano. Se hace inmediatamente después del nacimiento, en los primeros días de vida. Consiste en eliminar el ductus y seccionar el tronco de la arteria pulmonar, suturando su cabo distal (Dibujo linea superior izquierda). El cabo proximal se anastomosa con la aorta ascendente hipoplásica, el arco aórtico y la aorta descendente (Dibujo linea superior central) ampliando dicha anastomosis con un gran parche de pericardio (Dibujo linea superior derecha). El cabo distal de la arteria pulmonar junto con las ramas pulmonares queda independiente de la circulación entre VD, arteria pulmonar proximal y aorta. La realización de una fístula entre la aorta o una de sus arterias y la arteria pulmonar derecha viabilizar la circulación sanguínea hacia los pulmones para su oxigenación (Dibujo linea inferior izquierda). Esta es la técnica de la Operación de Norwood. Una variante de esta técnica es la Operación de Sano en la que se sustituye la fístula de Blalock Taussig por el implante de un conducto de goretex desde ventrículo derecho al cabo distal del tronco de la arteria pulmonar (Dibujo linea inferior derecha). Ambas variantes viabilizan la circulación pulmonar, evitando que el flujo hacia los pulmones no sea excesivo. En esta misma cirugía se amplía la CIA para que tenga un tamaño grande.
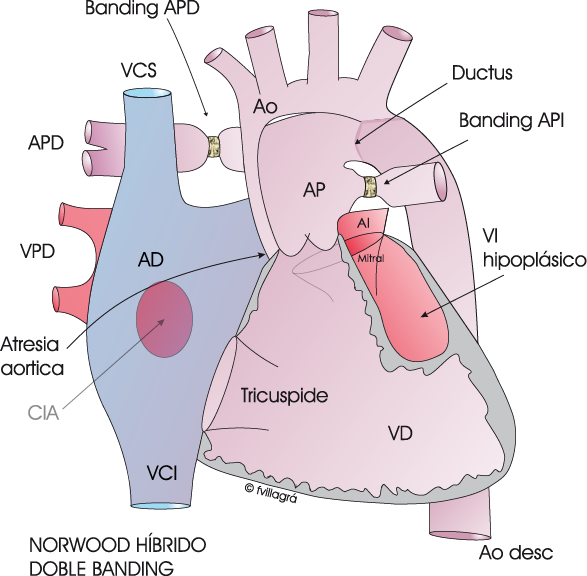

Norwood híbrido que está indicado en aquellos casos en los que hay firmes esperanzas de que el VI pudiera crecer y desarrollarse y conseguir una reparación quirúrgica final biventricular. El Norwood híbrido en estos casos proporciona tiempo para ver qué pasa con el VI. Consiste en realizar un Banding bilateral de las arterias pulmonares (Dibujo superior izquierdo) a través de una pequeña esternotomía media sin bypass cardiovascular, colocación de un stent en el ductus (Dibujo superior derecho) para que no se cierre y días después si necesario, la ampliación de la CIA o incluso implante de un stent para mantenerla bien amplia.

En las imágenes ecocardiográficas podemos observar varios fotogramas ecocardiográficos del Norwood híbrido. En las dos imágenes de la izquierda podemos visualizar el stent ductal con un flujo de la arteria pulmonar a la aorta, en la del centro el banding de la arteria pulmonar derecha y en la de la derecha un stent en la CIA para mantenerla amplia.
El manejo postquirúrgico no es sencillo y exige una unidad de intensivos muy especializada. Se debe controlar o evitar la parada circulatoria, de frecuencia nada desdeñable, el hiperaflujo pulmonar y su sobre-circulación, y ofrecer soporte circulatorio y respiratorio adecuado. Es fundamental una completa monitorización. La terapia médica coadyuvante incluye fármacos vasoactivos e inotrópicos, fármacos de sedo-analgesia, cierre de esternón diferido, y si necesario, el ECMO.
Los resultados han mejorado de manera importante durante las últimas 3 décadas, debido en parte a la mejora de los cuidados perioperatorios. Muchos de los estudios recientes aportan una mejoría de la supervivencia, arrojando unas tasas de supervivencia hospitalaria entre 74-93%. Múltiples estudios comunican una necesidad de reanimación cardiovascular con masaje entre 10-17%, y rescate en ECMO entre un 7-10%.
Periodo inter-estadío
A continuación de un exitoso Estadío-1 de paliación, hasta los supervivientes más estables y con mejores resultados corren riesgo de descompensación hemodinámica durante el período interestadío (tiempo desde el alta hospitalaria tras el Estadío-1 hasta el Estadío-II de paliación). La incidencia de mortalidad en esta etapa no es infrecuente, con unas tasas publicadas de un 2 a un 16%. Por lo tanto, en los pacientes con alta domiciliaria tras completar el primer Estadio de paliación, se debe garantizar un seguimiento estrecho durante el período de Norwood a Glenn. No es infrecuente que en este periodo se necesiten procedimientos percutáneos para permeabilizar una fístula que se haya obstruido o una conexión que se ha estenosado, o simplemente corregir estenosis en las arterias pulmonares o en la misma aorta.
Estadío-II de Paliación: Glenn

El Estadío-II de paliación es la conversión de un sistema de circulación pulmonar de alta presión (ventrículo derecho o aorta) a una fuente venosa de flujo pulmonar u operación de Glenn. Se realiza aproximadamente a los 6 meses de edad, y consiste en la realización de una anastomosis entre la vena cava superior y las arterias pulmonares, tal como indica el dibujo superior. Esta conexión cavopulmonar constituye una etapa fundamental, porque elimina del corazón la sangre que proviene de la vena cava superior, que ahora va directamente a los pulmones sin pasar por el corazón. Este ya no tiene que trabajar tanto, ya que no tiene que enviar esta sangre hacia los pulmones. (Ver ventrículo único). La mortalidad del estadío-II es baja, siendo la mortalidad hospitalaria virtual de cero, y la supervivencia al año de un 95%.
Estadío-III de Paliación (Fontan)

Se realiza a partir de los dos años de vida y consiste en la anastomosis de la vena cava inferior con las arterias pulmonares (operación de Fontan), que se realiza mediante un tubo extracardiaco como se ve en el dibujo superior o un parche interauricular, dependiendo de la edad y tamaño del paciente. Esta operación hace que el corazón trabaje todavía menos, pues toda la sangre que va a los pulmones lo hace de forma directa, sin pasar por el corazón. El corazón, libre de la sangre (azul) que tiene que ir a los pulmones, se encarga sólo de la que tiene que ir a todo el cuerpo, (roja) y trabaja normalmente preservando su durabilidad. Esta operación separa definitivamente los dos tipos de sangre, la Azul y la Roja, y permite eliminar la cianosis. Ver ventrículo único.
Trasplante Cardiaco
El trasplante cardiaco puede ser utilizado como terapia de rescate en cualquiera de los estadios de paliación de SCIH, y más frecuentemente tras completar el Fontan para todas las fisiologías univentriculares. No existe un consenso claro de cuál es el momento más propicio para decidir listar para trasplante cardiaco a un paciente con SCIH. Es importante saber que los pacientes con fisiología univentricular son particularmente difíciles de mantener en una asistencia circulatoria prolongada mientras esperan en lista de trasplante cardiaco, con medidas de ventilación mecánica, dosis altas de inotrópicos, etc, y no existe una asistencia ventricular ideal como puente al trasplante (ECMO o Berlin Heart).