 Cardiopatías Congénitas
Cardiopatías CongénitasLa atresia pulmonar con comunicación interventricular (AP CIV) representa la forma extrema de la Tetralogía de Fallot. Puede ser una lesión aislada o asociada a un síndrome genético. Se ha demostrado que la Tetralogía de Fallot con Atresia Pulmonar pertenece a un espectro de malformaciones cardiacas cono-truncales que se pueden asociar con monosomías 22q11. La presentación clínica de la monosomía 22q11 incluye pacientes con síndromes de línea media, Síndrome velo-cardio-facial y síndrome de DiGeorge. Más recientemente, estos síndromes se han incorporado como grupo bajo el acrónimo CATCH 22 (defecto cardíaco, anomalías craneales, hipoplasia tímica, paladar hendido, hipocalcemia y microdeleción 22q11).
En la atresia pulmonar con comunicación interventricular (AP CIV) no hay conexión anatómica entre el ventrículo derecho y las arterias pulmonares: Están ausentes 1) la válvula pulmonar en todos los casos, 2) el tronco pulmonar en la mayoría y 3) en algunos pocos hay ausencia también de la porción del ventrículo derecho (infundíbulo) más cercana a las arterias pulmonares.
La sangre no oxigenada (azul) del ventrículo derecho no puede salir hacia los pulmones (P) a oxigenarse, debido a la ausencia de conexión anatómica entre el ventrículo derecho y las arterias pulmonares tal como se ve en el esquema superior. Esta sangre se desvía hacia la aorta a través de la CIV. La aorta recibe la escasa sangre oxigenada (roja) del ventrículo izquierdo (escasa porque es la que procede del pulmón, que a su vez es escasa porque no hay conexión entre el ventrículo derecho y los pulmones), y la sangre no oxigenada (azul) del ventrículo derecho que pasa por la CIV mezclándose y resultando al final en una sangre de escasa oxigenación (violeta), que ocasiona el color azulado (cianosis) de estos niños, al distribuirse por todo el cuerpo (C) o circulación sistémica.
Esta situación, en la que no pasa sangre alguna hacia los pulmones para oxigenarse, es incompatible con la vida y estos niños fallecerían nada más nacer. Viven porque la sangre llega a los pulmones no por su sitio, pero sí por vías anómalas. Hay varios tipos de vías anómalas:
Tipo I: 40% de los casos de AP CIV

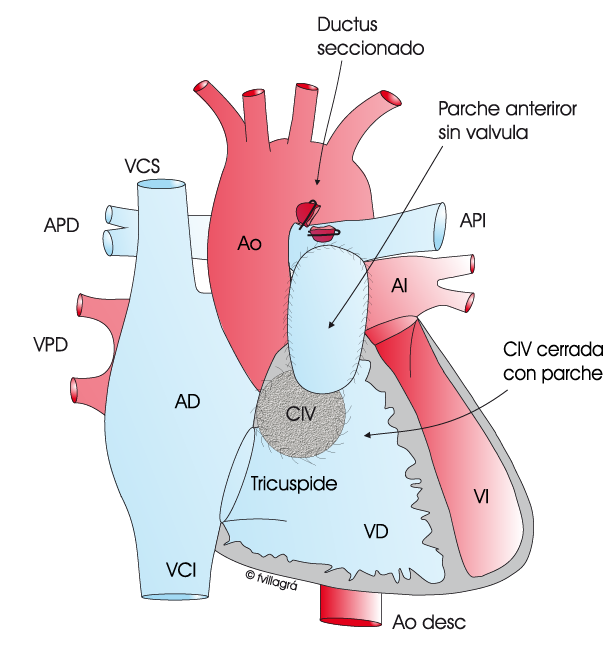
La primera vía anómala es el ductus que está abierto y permite el paso de sangre mezclada y pobremente oxigenada (violeta) desde la aorta a la arteria pulmonar (AP), tal como se ve en la figura superior izquierda y en el esquema circulatorio animado superior. El pulmón (P) recibe esta sangre mezclada (violeta) oxigenando la porción no oxigenada. Desde el pulmón sale ya la sangre totalmente oxigenada (roja) por las venas pulmonares derechas e izquierdas (VPD y VPI) hacia la aurícula (AI) y ventrículo izquierdo (VI). En este tipo de vía lo normal es que ambas arterias pulmonares, la que va al pulmón derecho (arteria pulmonar derecha) (APD) y la que va al pulmón izquierdo (arteria pulmonar izquierda) (API) sean confluentes (están unidas), de forma que la sangre que proviene del ductus se distribuye homogéneamente entre ambas y sean de buen tamaño.
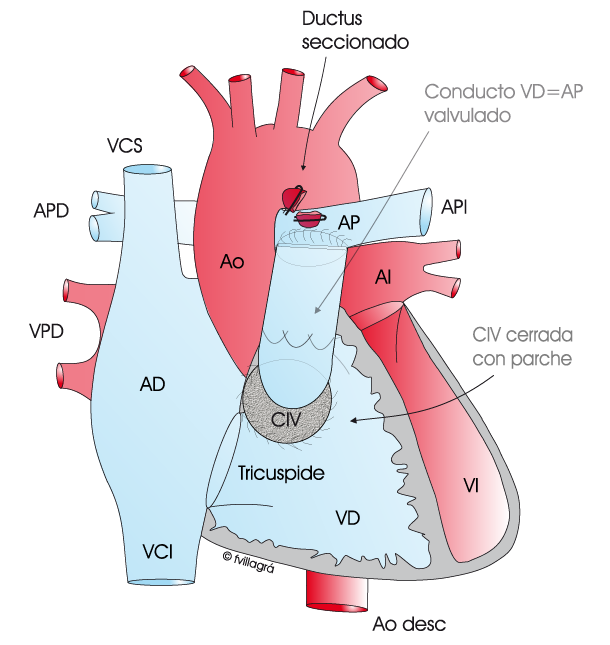
La cirugía consiste en 1) ocluir el ductus, 2) cerrar la CIV con parche y 3) conectar el ventrículo derecho a las arterias pulmonares. Hay varias técnicas para realizar esta conexión. La ideal es la unión directa de las arterias pulmonares en su cara posterior al ventrículo derecho, ampliando la cara anterior con un parche tal como se puede ver en la figura superior central; en general es una conexión duradera, no siendo necesarias las reoperaciones; sin embargo, esta técnica no proporciona una válvula pulmonar, por lo que es inevitable que exista en estos niños insuficiencia valvular pulmonar en grado variable, que a largo plazo requerirá el implante de una prótesis valvular pulmonar. Si la distancia entre el ventrículo derecho y las arterias pulmonares es grande, la anterior técnica no es siempre factible y la conexión se realiza con un conducto avalvulado o valvulado tal como se ve en la figura superior derecha. El conducto valvulado (de procedencia animal o humana –homoinjerto-) proporciona una válvula pulmonar en su interior, y por lo tanto evita la insuficiencia valvular pulmonar; sin embargo, estos conductos valvulados se pueden quedar pequeños al crecer el niño y/o en un buen número de casos se deterioran con el tiempo, siendo necesario su reemplazo por otro, en una reoperación futura.
Tipo II: 25% de los casos de AP CIV
En este tipo, la vía anómala por la que llega sangre a los pulmonares es también y habitualmente por un ductus, pero las arterias pulmonares son pequeñas e incluso hipoplásicas.
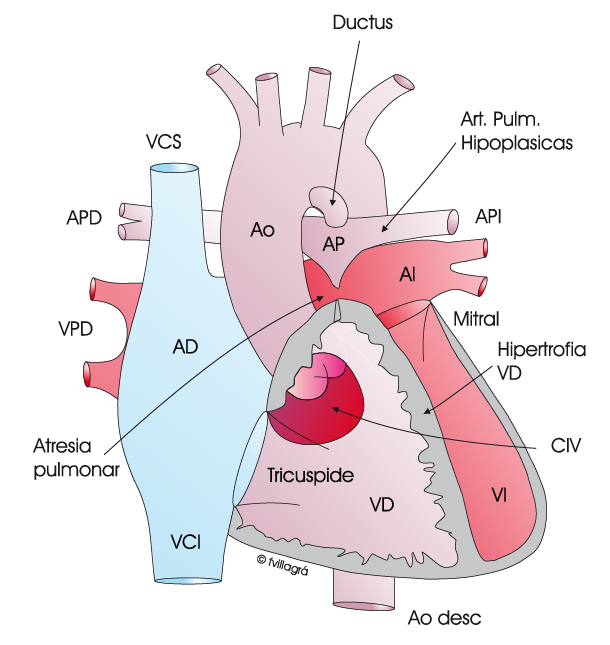
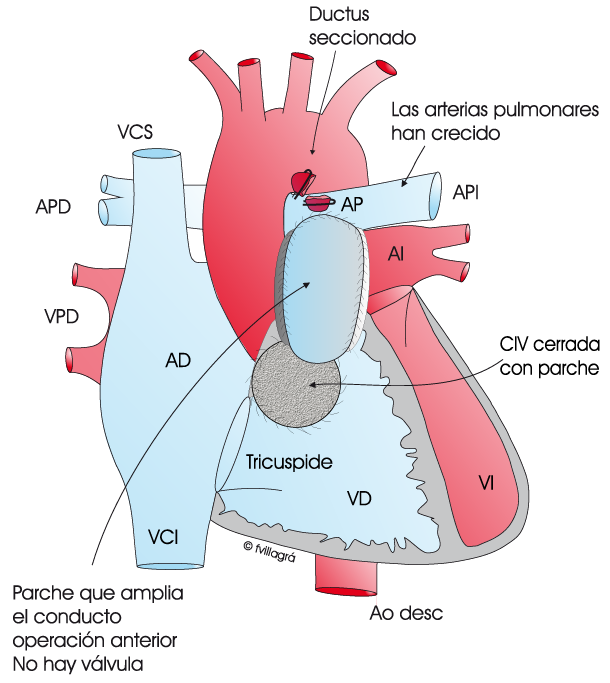
En la figura superior de la izquierda se representa este tipo de APCIV.
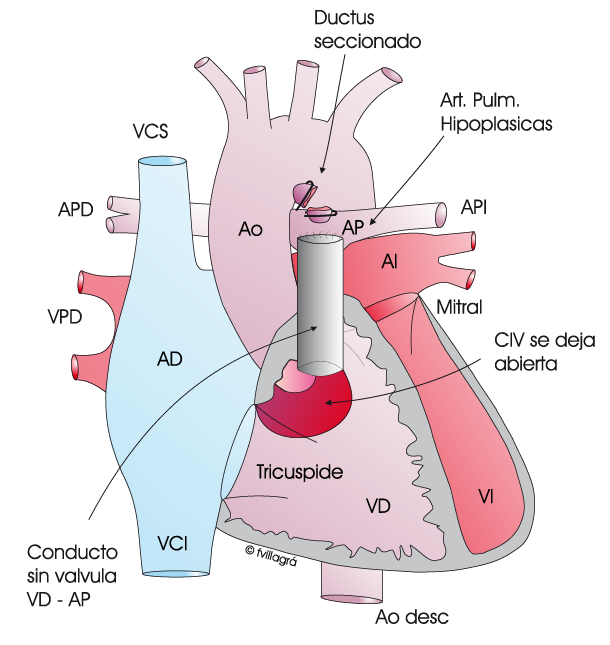
La cirugía consiste en 1) ocluir el ductus, 2) conectar el ventrículo derecho a las arterias pulmonares con un conducto no valvulado y 3) dejar la CIV abierta, tal como se observa en la figura superior central. Efectivamente, la pequeñez de las arterias pulmonares no admite todo el flujo normal de sangre hacia los pulmones y es necesario dejar la CIV abierta, para que parte de la sangre no oxigenada del ventrículo derecho «se escape» hacia el ventrículo izquierdo. Se trata de una operación paliativa, intermedia, también llamada “hemi-corrección”. Su objetivo es propiciar el crecimiento de estas arterias pulmonares pequeñas mediante el propio paso de la sangre. En estos casos, el tipo de conexión ventrículo derecho-arterias pulmonares es un parche o un conducto avalvulado algo más pequeño que el que correspondería por edad. Cuando las arterias pulmonares hayan adquirido un tamaño normal, se realiza la operación definitiva consistente en el cierre de la CIV y ampliación del conducto o parche (figura superior derecha) para que la conexión entre ventrículo derecho y arterias pulmonares tenga un diámetro normal.
Tipo III: 15% de los casos de AP CIV
La vía anómala de flujo a los pulmones es un ductus o colaterales pero además las dos arterias pulmonares, la que va al pulmón derecho y la que va al pulmón izquierdo, no son confluentes (no están unidas) y nacen cada una y reciben sangre de un sitio distinto. Estos casos son más complicados. La cirugía incluye todo lo expuesto en el tipo I (si las arterias pulmonares son de buen tamaño) o en el tipo II (si las arterias pulmonares son de pequeño tamaño), y además es necesario realizar la unión de ambas arterias pulmonares con anastomosis directas o interponiendo un conducto avalvulado o estructura similar.
Tipo IV: 15-20% de los casos de AP CIV
En este tipo la vía anómala de flujo sanguineo hacia los pulmones consiste en colaterales sistémico-pulmonares, que en inglés son conocidas como MAPCAS. Las MAPCAS permiten el paso de sangre mezclada y pobremente oxigenada (violeta) desde la aorta al pulmón (P) directamente tal como se observa en el esquema animado inferior. El pulmón (P) recibe esta sangre mezclada (violeta) oxigenando la porción no oxigenada. Desde el pulmón sale ya la sangre totalmente oxigenada (roja) por las venas pulmonares (VP) hacia la aurícula (AI) y ventrículo izquierdo (VI). En estos casos no hay arterias pulmonares verdaderas. Las MAPCAS son vasos tortuosos, presentan en su mayoría zonas estrechas, o por el contrario permiten el paso torrencial de sangre hacia los pulmones. Estos casos presentan una patología compleja y difícil de corregir.
Modernamente se realiza en ellos unifocalización de las MAPCAS, que consiste en unir todas las MAPCAS o colaterales en una estructura vascular común a base de anastomosis múltiples entre ellas tal como se observa en los dibujos; esta estructura común haría las veces de arterias pulmonares verdaderas: A esta estructura vascular se llevaría sangre desde el ventrículo derecho mediante conexión con parches o conductos.

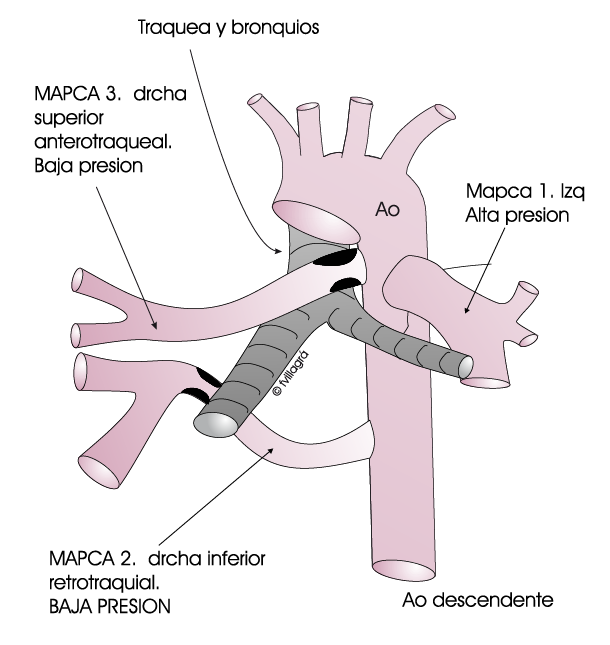
La CIV se podría cerrar dependiendo del volumen pulmonar irrigado por las MAPCAS unifocalizadas y/o por el diámetro de la estructura vascular común. Si el volumen pulmonar o el diámetro son pequeños, hay que posponer el cierre de la CIV para otra operación. En estos casos son frecuentes los cateterismos terapéuticos cuyo objetivo es ampliar zonas arteriales pulmonares estrechas con dilatación o implantes de “stents”, ocluir colaterales no accesibles quirúrgicamente, etc.
No son infrecuentes los casos con combinación de varios tipos de vías pulmonares anómalas: Casos con arterias pulmonares hipoplásicas y MAPCAS, casos con arterias pulmonares normales y con 1-2 MAPCAS, etc. Estos casos requieren actuaciones específicas.
En los tipos II, III y IV, la acción terapéutica médica combinada (cateterismo y cirugía) puede ser compleja, múltiple y prolongada en el tiempo. Esta acción terapéutica recibe el nombre de “rehabilitación de las arterias pulmonares”. La técnica consiste en 1) cierre de la CIV, 2) conexión VD-AP y/o 3) unifocalización de todas las MAPCAS. A pesar de ella, no en todos los niños se logra al final del proceso la corrección total de esta patología. En todos los 4 tipos, incluso con corrección total, queda insuficiencia valvular pulmonar de grado variable (si no hay válvula pulmonar implantada) que requerirá a largo plazo el implante de una prótesis valvular pulmonar, o posibilidad constante de reoperación para sustituir el conducto implantado (en los casos con un conducto valvulado).
Planificación del tratamiento quirúrgico:
La planificación del tratamiento quirúrgico debe ser adaptado a cada caso, pero debe tener en cuenta ciertos aspectos básicos:
Entre 1 y 6 meses de edad hay que asegurar un flujo pulmonar adecuado en cantidad y presión, así como un crecimiento de las arterias pulmonares en los casos en que son hipoplásicas. En los casos con flujo pulmonar escaso y/o con arterias pulmonares hipoplásicas, hay que realizar fístulas sistémico-pulmonares tipo Blalock-Taussig o conexión del ventrículo derecho con las arterias pulmonares sin cerrar la comunicación interventricular (hemicorrecciones). En algunos casos con flujo pulmonar excesivo o con altas presiones, hay que unifocalizar parcial y precozmente las MAPCAS sin estenosis para evitar la hipertensión pulmonar. Los casos tipo I pueden ser sometidos a reparación total, incluyendo el cierre de la comunicación interventricular. En los casos del tipo IV puede ser una alternativa quirúrgica unifocalizar las MAPCAS de un solo lado por toracotomía.
Entre los 6 meses y 12 meses de edad: Considerar la unifocalización completa con o sin cierre de la comunicación interventricular en los casos con MAPCAS. En los casos del tipo IV puede ser una alternativa quirúrgica unifocalizar las MAPCAS del lado restante por toracotomía.
Entre los 12 y 24 meses o incluso más tarde: Considerar el cierre de la comunicación interventricular en los casos con arterias pulmonares con Índice de McGoon y Nakata adecuados o en aquellos casos en los que se ha procedido a la unifocalización y tienen un desarrollo del árbol pulmonar suficiente.
Cuando sea necesario en el futuro: Implante de una prótesis valvular pulmonar antes de que el ventrículo derecho se dilate en exceso.
El cateterismo diagnóstico y terapéutico es constantemente necesario a lo largo del tratamiento quirúrgico.